To illustrate the functionality of the dar package, this
study will use the data set from (Noguera-Julian, M., et al. 2016). The
authors of this study found that men who have sex with men (MSM)
predominantly belonged to the Prevotella-rich enterotype whereas most
non-MSM subjects were enriched in Bacteroides, independently of HIV-1
status. This result highlights the potential impact of sexual
orientation on the gut microbiome and emphasizes the importance of
controlling for such variables in microbiome research. Using the
dar package, we will conduct a differential abundance
analysis to further explore this finding and uncover potential microbial
biomarkers associated with this specific population.
Load dar package and data
library(dar)
# suppressPackageStartupMessages(library(plotly))
data("metaHIV_phy")
metaHIV_phy
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 451 taxa and 156 samples ]
#> sample_data() Sample Data: [ 156 samples by 3 sample variables ]
#> tax_table() Taxonomy Table: [ 451 taxa by 7 taxonomic ranks ]Recipe initialization
To begin the analysis process with the dar package, the
first step is to initialize a Recipe object, which is an S4
class. This recipe object serves as a blueprint for the data preparation
steps required for the differential abundance analysis. The
initialization of the recipe object is done through the function
recipe(), which takes as inputs a phyloseq or
TreeSummarizedExperiment (TSE) object, the name of the
categorical variable of interest and the taxonomic level at which the
differential abundance analyses are to be performed. As previously
mentioned, we will use the data set from (Noguera-Julian, M., et
al. 2016) and the variable of interest “RiskGroup2” containing the
categories: men who have sex with men (msm), non-MSM (hts) and people
who inject drugs (pwid) and we will perform the analysis at the species
level.
## Recipe initialization
rec <- recipe(metaHIV_phy, var_info = "RiskGroup2", tax_info = "Species")
rec
#> ── DAR Recipe ──────────────────────────────────────────────────────────────────
#> Inputs:
#>
#> ℹ phyloseq object with 451 taxa and 156 samples
#> ℹ variable of interes RiskGroup2 (class: character, levels: hts, msm, pwid)
#> ℹ taxonomic level SpeciesRecipe QC and preprocessing steps definition
Once the recipe object has been initialized, the next step is to
populate it with steps. Steps are the methods that will be applied to
the data stored in the recipe. There are two types of steps:
preprocessing (prepro) and differential abundance (da) steps. Initially,
we will focus on the prepro steps which are used to modify the data
loaded into the recipe, which will then be used for the da steps. The
‘dar’ package includes 3 main preprocessing functionalities:
step_subset_taxa, which is used for subsetting columns and
values in the taxon table connected to the phyloseq object,
step_filter_taxa, which is used to filter the OTUs, and
step_rarefaction, which is used to resample the OTU table
to ensure that all samples have the same library size. These
functionalities allow for a high level of flexibility and customization
in the data preparation process before performing the differential
abundance analysis.
The dar package provides convenient wrappers for the
step_filter_taxa function, designed to filter Operational
Taxonomic Units (OTUs) based on specific criteria: prevalence, variance,
abundance, and rarity.
-
step_filter_by_prevalence: Filters OTUs according to the number of samples in which the OTU appears. -
step_filter_by_variance: Filters OTUs based on the variance of the OTU’s presence across samples. -
step_filter_by_abundance: Filters OTUs according to the OTU’s abundance across samples. -
step_filter_by_rarity: Filters OTUs based on the rarity of the OTU across samples.
In addition to the preprocessing steps, the dar package
also incorporates the function phy_qc which returns a table
with a set of metrics that allow for informed decisions to be made about
the data preprocessing that will be done. In our case, we decided to use
the step_subset_taxa function to retain only those observations
annotated within the realm of Bacteria and Archaea. We also used the
step_filter_by_prevalence function to retain only those
OTUs with at least 1% of the samples with values greater than 0. This
approach ensured that we were working with a high-quality, informative
subset of the data, which improved the overall accuracy and reliability
of the differential abundance analysis.
## QC
phy_qc(rec)
#> # A tibble: 4 × 12
#> var_levels n n_zero pct_zero pct_all_zero pct_singletons pct_doubletons
#> <chr> <int> <int> <dbl> <dbl> <dbl> <dbl>
#> 1 all 70356 57632 81.9 0 20.6 8.87
#> 2 hts 18491 15108 81.7 24.2 22.8 8.43
#> 3 msm 45100 37019 82.1 16.0 20.2 9.53
#> 4 pwid 6765 5505 81.4 41.2 16.6 9.31
#> # ℹ 5 more variables: n_samples <int>, lib_size_min <dbl>, lib_size_max <dbl>,
#> # count_mean <dbl>, count_max <dbl>
## Adding prepro steps
rec <-
rec |>
step_subset_taxa(tax_level = "Kingdom", taxa = c("Bacteria", "Archaea")) |>
step_filter_by_prevalence()
rec
#> ── DAR Recipe ──────────────────────────────────────────────────────────────────
#> Inputs:
#>
#> ℹ phyloseq object with 451 taxa and 156 samples
#> ℹ variable of interes RiskGroup2 (class: character, levels: hts, msm, pwid)
#> ℹ taxonomic level Species
#>
#> Preporcessing steps:
#>
#> ◉ step_subset_taxa() id = subset_taxa__Puff_pastry
#> ◉ step_filter_by_prevalence() id = filter_by_prevalence__Paris–Brest
#>
#> DA steps:Define Differential Analysis (DA) steps
Once data is preprocessed and cleaned, the next step is to add the da
steps. The dar package incorporates multiple methods to
analyze the data, including: ALDEx2, ANCOM-BC, corncob, DESeq2, Lefse,
MAaslin3 and Wilcox. These methods provide a range of options for
uncovering potential microbial biomarkers associated with the variable
of interest. To ensure consistency across methods, we decided not to use
default parameters, but to set the min_prevalence parameter
to 0 for MAaslin3. This approach ensured that the analysis was
consistent across all methods and that the results were
interpretable.
Note: to reduce computation time, in this example we will only use the DESeq2 and MAaslin3 methods, that are the fastest ones. However, we recommend using all the methods available in the package to ensure a more robust analysis.
## DA steps definition
rec <- rec |>
step_deseq() |>
step_maaslin(min_prevalence = 0)
rec
#> ── DAR Recipe ──────────────────────────────────────────────────────────────────
#> Inputs:
#>
#> ℹ phyloseq object with 451 taxa and 156 samples
#> ℹ variable of interes RiskGroup2 (class: character, levels: hts, msm, pwid)
#> ℹ taxonomic level Species
#>
#> Preporcessing steps:
#>
#> ◉ step_subset_taxa() id = subset_taxa__Puff_pastry
#> ◉ step_filter_by_prevalence() id = filter_by_prevalence__Paris–Brest
#>
#> DA steps:
#>
#> ◉ step_deseq() id = deseq__Spina_santa
#> ◉ step_maaslin() id = maaslin__Dutch_Baby_PancakePrep recipe
Once the recipe has been defined, the next step is to execute all the
steps defined in the recipe. This is done through the function
prep(). Internally, it first executes the preprocessing
steps, which modify the phyloseq object stored in the recipe. Then,
using the modified phyloseq, it executes each of the defined
differential abundance methods. To speed up the execution time, the
prep() function includes the option to run in parallel. The
resulting object has class PrepRecipe and when printed in
the terminal, it displays the number of taxa detected as significant in
each of the methods and also the total number of taxa shared across all
methods. This allows for a provisional overview of the results and a
comparison between methods.
## Execute in parallel
da_results <- prep(rec, parallel = TRUE)
da_results
#> ── DAR Results ─────────────────────────────────────────────────────────────────
#> Inputs:
#>
#> ℹ phyloseq object with 355 taxa and 156 samples
#> ℹ variable of interes RiskGroup2 (class: character, levels: hts, msm, pwid)
#> ℹ taxonomic level Species
#>
#> Results:
#>
#> ✔ deseq__Spina_santa diff_taxa = 174
#> ✔ maaslin__Dutch_Baby_Pancake diff_taxa = 58
#>
#> ℹ 31 taxa are present in all tested methodsDefault results extraction
At this point, we could extract the taxa shared across all methods
using the function bake() to define a default consensus
strategy and then cool() to extract the results.
## Default DA taxa results
results <-
bake(da_results) |>
cool()
results
#> # A tibble: 31 × 2
#> taxa_id taxa
#> <chr> <chr>
#> 1 Otu_35 Collinsella_aerofaciens
#> 2 Otu_37 Collinsella_stercoris
#> 3 Otu_47 Bacteroides_cellulosilyticus
#> 4 Otu_48 Bacteroides_clarus
#> 5 Otu_63 Bacteroides_plebeius
#> 6 Otu_69 Bacteroides_sp_CAG_530
#> 7 Otu_78 Bacteroides_uniformis
#> 8 Otu_82 Barnesiella_intestinihominis
#> 9 Otu_96 Prevotella_copri
#> 10 Otu_102 Prevotella_sp_AM42_24
#> # ℹ 21 more rowsHowever, dar allows for complex consensus strategies
based on the obtained results. To that end, the user has access to
different functions to graphically represent different types of
information. This feature allows for a more in-depth analysis of the
results and a better understanding of the underlying patterns in the
data.
Exploration for consensus strategie definition
For example, intersection_plt() gives an overview of the
overlaps between methods by creating an upSet plot. In our case, this
function has shown that 210 taxa are shared across all the methods
used.
## Intersection plot
intersection_plt(da_results, ordered_by = "degree", font_size = 1)
#> Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
#> ℹ Please use tidy evaluation idioms with `aes()`.
#> ℹ See also `vignette("ggplot2-in-packages")` for more information.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue at <https://github.com/hms-dbmi/UpSetR/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue at <https://github.com/hms-dbmi/UpSetR/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
#> Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
#> ℹ Please use the `linewidth` argument instead.
#> ℹ The deprecated feature was likely used in the UpSetR package.
#> Please report the issue at <https://github.com/hms-dbmi/UpSetR/issues>.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.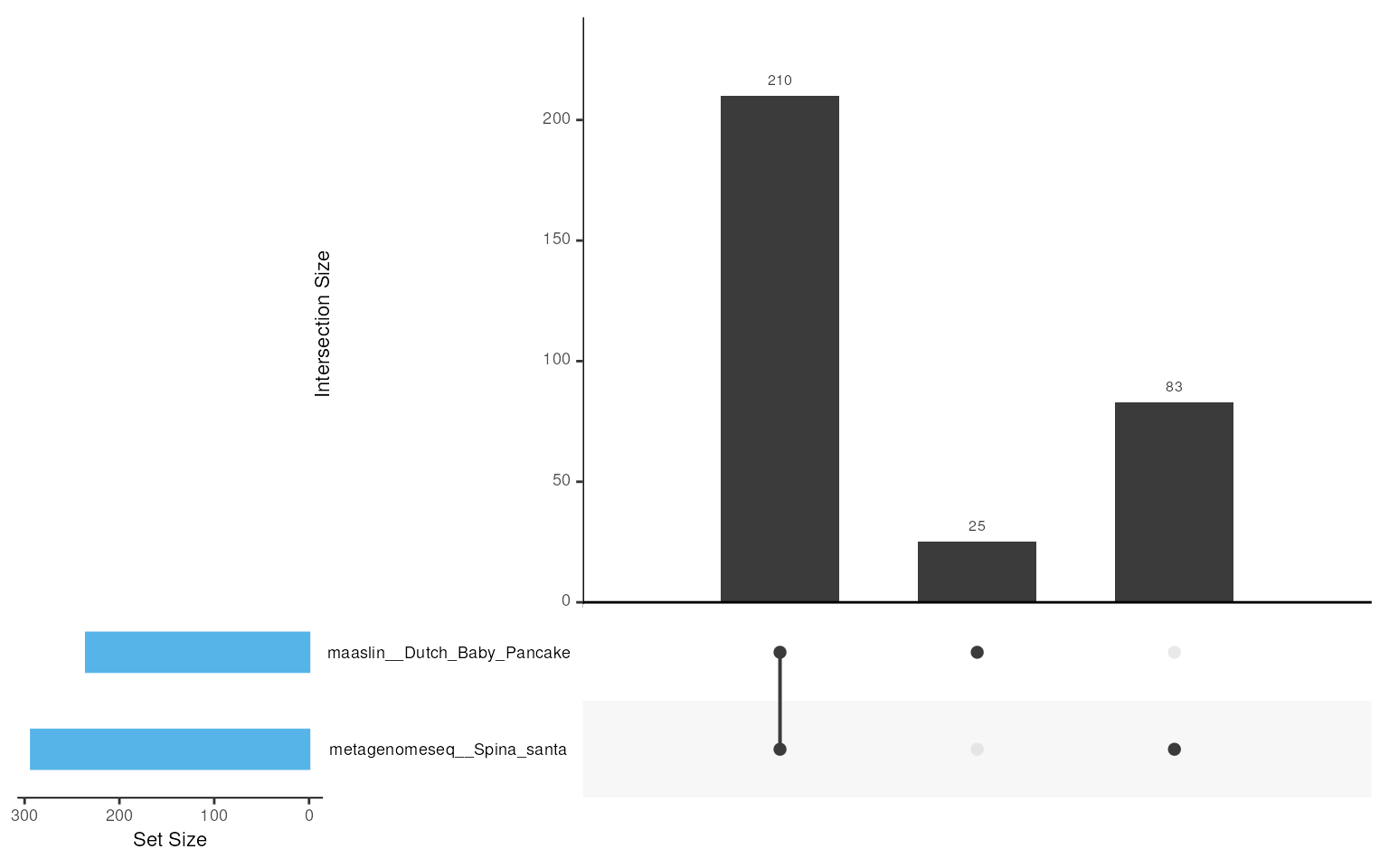
In addition to the intersection_plt() function,
dar also has the function exclusion_plt()
which provides information about the number of OTUs shared between
methods. This function allows to identify the OTUs that are specific to
each method and also the ones that are not shared among any method.
## Exclusion plot
exclusion_plt(da_results)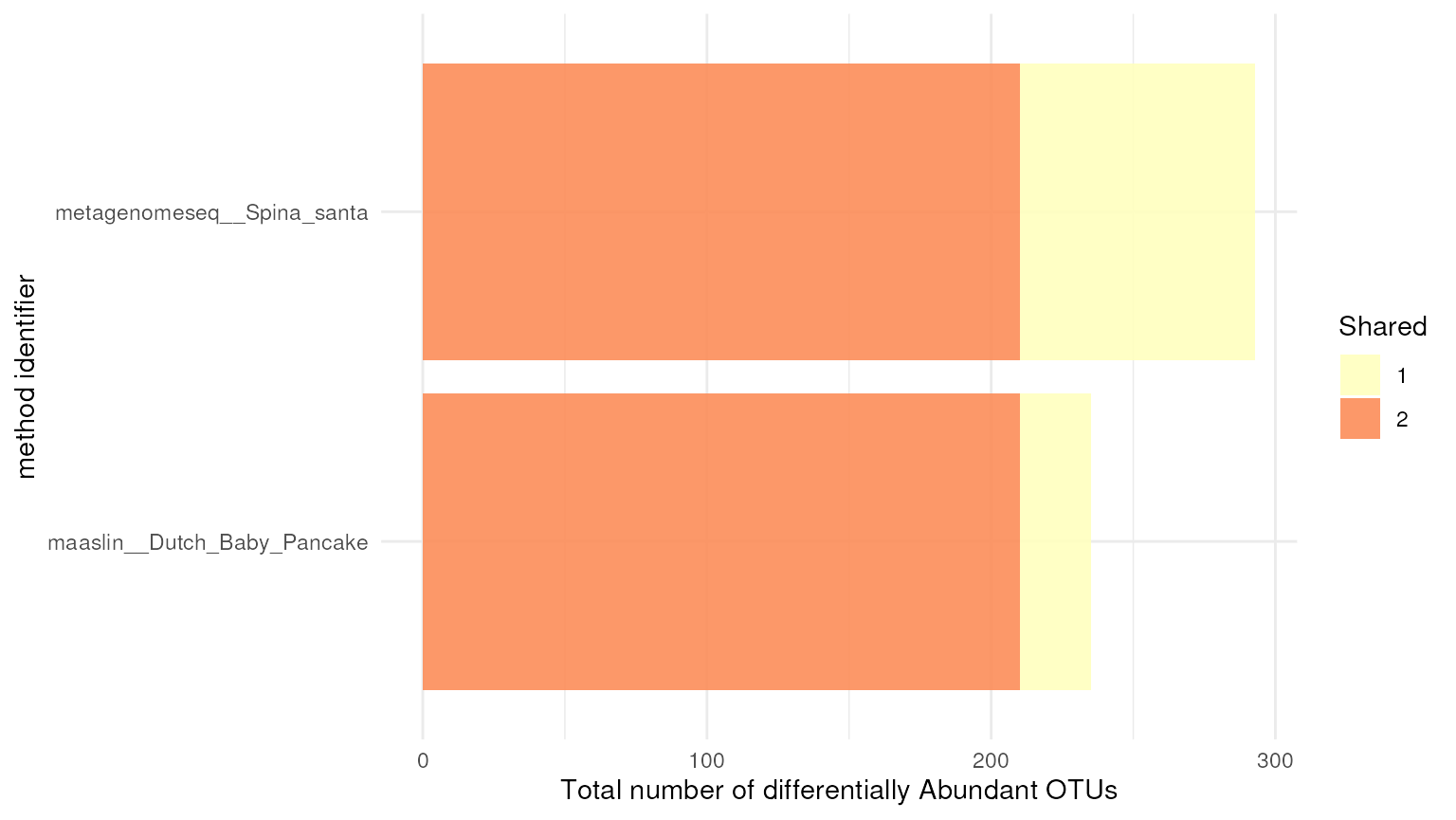
Besides to the previously mentioned functions, dar also
includes the function corr_heatmap(), which allows for
visualization of the overlap of significant OTUs between tested methods.
This function can provide similar information to the previous plots, but
in some cases it may be easier to interpret. comprehensive view of the
results.
## Correlation heatmap
corr_heat <- corr_heatmap(da_results, font_size = 10)
corr_heatFinally, dar also includes the function
mutual_plt(), which plots the number of differential
abundant features mutually found by a defined number of methods, colored
by the differential abundance direction and separated by comparison. The
resulting graph allows us to see that the features detected correspond
mainly to the comparisons between hts vs msm and msm vs pwid.
Additionally, the graph also allows us to observe the direction of the
effect; whether a specific OTU is enriched or depleted for each
comparison.
## Mutual plot
mutual_plt(
da_results,
count_cutoff = length(steps_ids(da_results, type = "da"))
)
Define a consesus strategy using bake
After visually inspecting the results from running all the
differential analysis methods on our data, we have the necessary
information to define a consensus strategy that fits our dataset. In our
case, we will retain all the methods. However if one or more methods are
not desired, the bake() function includes the
exclude parameter, which allows to exclude specific
methods.
Additionally, the bake() function allows to further
refine the consensus strategy through its parameters, such as
count_cutoff, which indicates the minimum number of methods
in which an OTU must be present, and weights, a named
vector with the ponderation value for each method. However, for
simplicity, these parameters are not used in this example.
## Define consensus strategy
da_results <- bake(da_results)
da_results
#> ── DAR Results ─────────────────────────────────────────────────────────────────
#> Inputs:
#>
#> ℹ phyloseq object with 355 taxa and 156 samples
#> ℹ variable of interes RiskGroup2 (class: character, levels: hts, msm, pwid)
#> ℹ taxonomic level Species
#>
#> Results:
#>
#> ✔ deseq__Spina_santa diff_taxa = 174
#> ✔ maaslin__Dutch_Baby_Pancake diff_taxa = 58
#>
#> ℹ 31 taxa are present in all tested methods
#>
#> Bakes:
#>
#> ◉ 1 -> count_cutoff: NULL, weights: NULL, exclude: NULL, id: bake__Cornish_pastyExtract results
To conclude, we can extract the final results using the
cool() function. This function takes a
PrepRecipe object and the ID of the bake to be used as
input (by default it is 1, but if you have multiple consensus
strategies, you can change it to extract the desired results).
## Extract results for bake id 1
f_results <- cool(da_results, bake = 1)
f_results
#> # A tibble: 31 × 2
#> taxa_id taxa
#> <chr> <chr>
#> 1 Otu_35 Collinsella_aerofaciens
#> 2 Otu_37 Collinsella_stercoris
#> 3 Otu_47 Bacteroides_cellulosilyticus
#> 4 Otu_48 Bacteroides_clarus
#> 5 Otu_63 Bacteroides_plebeius
#> 6 Otu_69 Bacteroides_sp_CAG_530
#> 7 Otu_78 Bacteroides_uniformis
#> 8 Otu_82 Barnesiella_intestinihominis
#> 9 Otu_96 Prevotella_copri
#> 10 Otu_102 Prevotella_sp_AM42_24
#> # ℹ 21 more rowsTo further visualize the results, the abundance_plt()
function can be utilized to visualize the differences in abundance of
the differential abundant taxa.
## Ids for Bacteroide and Provotella species
ids <-
f_results |>
dplyr::filter(stringr::str_detect(taxa, "Bacteroi.*|Prevote.*")) |>
dplyr::pull(taxa_id)
## Abundance plot as boxplot
abundance_plt(da_results, taxa_ids = ids, type = "boxplot") 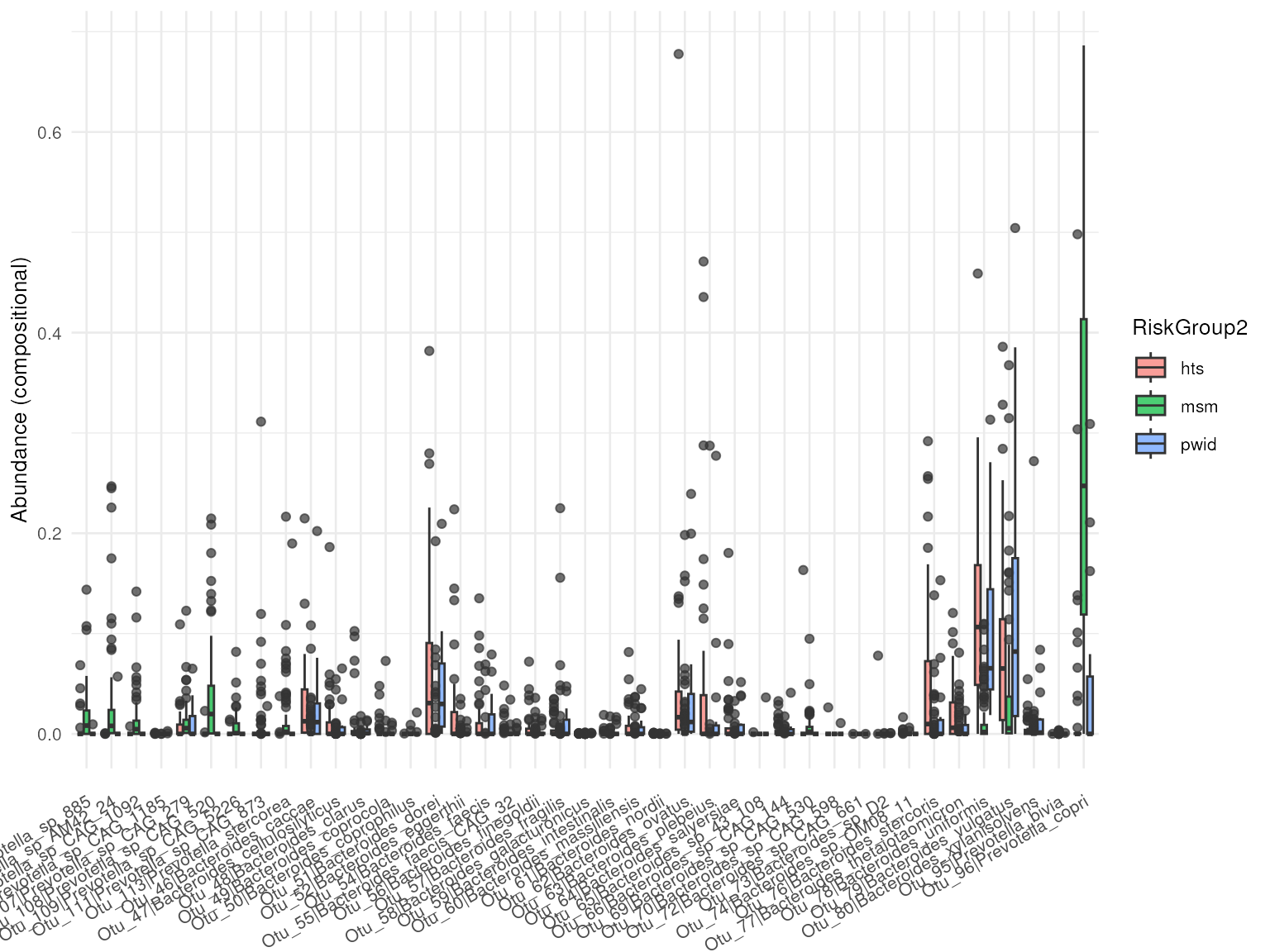
Session info
devtools::session_info()
#> ─ Session info ───────────────────────────────────────────────────────────────
#> setting value
#> version R version 4.6.1 (2026-06-24)
#> os Ubuntu 24.04.4 LTS
#> system x86_64, linux-gnu
#> ui X11
#> language en
#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
#> date 2026-07-29
#> pandoc 3.10 @ /usr/bin/ (via rmarkdown)
#> quarto 1.9.38 @ /usr/local/bin/quarto
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
#> abind 1.4-8 2024-09-12 [1] RSPM (R 4.6.0)
#> ade4 1.7-24 2026-03-21 [1] RSPM (R 4.6.0)
#> ape 5.8-1 2024-12-16 [1] RSPM (R 4.6.0)
#> apeglm 1.34.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> ashr 2.2-63 2023-08-21 [1] RSPM (R 4.6.0)
#> assertthat 0.2.1 2019-03-21 [1] RSPM (R 4.6.0)
#> backports 1.5.1 2026-04-03 [1] RSPM (R 4.6.0)
#> bbmle 1.0.25.1 2023-12-09 [1] RSPM (R 4.6.0)
#> bdsmatrix 1.3-7 2024-03-02 [1] RSPM (R 4.6.0)
#> beachmat 2.28.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> beeswarm 0.4.0 2021-06-01 [1] RSPM (R 4.6.0)
#> Biobase 2.72.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> BiocGenerics 0.58.1 2026-05-14 [1] Bioconductor 3.23 (R 4.6.1)
#> BiocNeighbors 2.6.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> BiocParallel 1.46.0 2026-04-29 [1] Bioconductor 3.23 (R 4.6.1)
#> BiocSingular 1.28.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> biomformat 1.40.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> Biostrings 2.80.1 2026-05-22 [1] Bioconductor 3.23 (R 4.6.1)
#> bluster 1.22.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> bslib 0.11.0 2026-05-16 [2] RSPM (R 4.6.0)
#> ca 0.71.1 2020-01-24 [1] RSPM (R 4.6.0)
#> cachem 1.1.0 2024-05-16 [2] RSPM (R 4.6.0)
#> checkmate 2.3.4 2026-02-03 [1] RSPM (R 4.6.0)
#> cli 3.6.6 2026-04-09 [2] RSPM (R 4.6.0)
#> cluster 2.1.8.2 2026-02-05 [3] CRAN (R 4.6.1)
#> coda 0.19-4.1 2024-01-31 [1] RSPM (R 4.6.0)
#> codetools 0.2-20 2024-03-31 [3] CRAN (R 4.6.1)
#> crayon 1.5.3 2024-06-20 [2] RSPM (R 4.6.0)
#> crosstalk 1.2.2 2025-08-26 [1] RSPM (R 4.6.0)
#> dar * 1.9.1 2026-07-29 [1] Bioconductor
#> data.table 1.18.4 2026-05-06 [1] RSPM (R 4.6.0)
#> DBI 1.3.0 2026-02-25 [1] RSPM (R 4.6.0)
#> DECIPHER 3.8.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> decontam 1.32.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> DelayedArray 0.38.2 2026-05-26 [1] Bioconductor 3.23 (R 4.6.1)
#> DelayedMatrixStats 1.34.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> dendextend 1.19.1 2025-07-15 [1] RSPM (R 4.6.0)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.6.0)
#> DESeq2 1.52.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> devtools 2.5.2 2026-04-30 [2] RSPM (R 4.6.0)
#> digest 0.6.39 2025-11-19 [2] RSPM (R 4.6.0)
#> DirichletMultinomial 1.54.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> dplyr 1.2.1 2026-04-03 [1] RSPM (R 4.6.0)
#> ecodive 2.2.6 2026-04-14 [1] RSPM (R 4.6.0)
#> ellipsis 0.3.3 2026-04-04 [2] RSPM (R 4.6.0)
#> emdbook 1.3.14 2025-07-23 [1] RSPM (R 4.6.0)
#> evaluate 1.0.5 2025-08-27 [2] RSPM (R 4.6.0)
#> farver 2.1.2 2024-05-13 [1] RSPM (R 4.6.0)
#> fastmap 1.2.0 2024-05-15 [2] RSPM (R 4.6.0)
#> foreach 1.5.2 2022-02-02 [1] RSPM (R 4.6.0)
#> fs 2.1.0 2026-04-18 [2] RSPM (R 4.6.0)
#> furrr 0.4.0 2026-03-31 [1] RSPM (R 4.6.0)
#> future 1.75.0 2026-07-20 [1] RSPM (R 4.6.0)
#> generics 0.1.4 2025-05-09 [1] RSPM (R 4.6.0)
#> GenomicRanges 1.64.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> ggbeeswarm 0.7.3 2025-11-29 [1] RSPM (R 4.6.0)
#> ggplot2 4.0.3 2026-04-22 [1] RSPM (R 4.6.0)
#> ggrepel 0.9.8 2026-03-17 [1] RSPM (R 4.6.0)
#> globals 0.19.1 2026-03-13 [1] RSPM (R 4.6.0)
#> glue 1.8.1 2026-04-17 [2] RSPM (R 4.6.0)
#> gridExtra 2.3.1 2026-06-25 [1] RSPM (R 4.6.0)
#> gtable 0.3.6 2024-10-25 [1] RSPM (R 4.6.0)
#> heatmaply 1.6.0 2025-07-12 [1] RSPM (R 4.6.0)
#> htmltools 0.5.9 2025-12-04 [2] RSPM (R 4.6.0)
#> htmlwidgets 1.6.4 2023-12-06 [2] RSPM (R 4.6.0)
#> httr 1.4.8 2026-02-13 [1] RSPM (R 4.6.0)
#> igraph 2.3.3 2026-06-26 [1] RSPM (R 4.6.0)
#> invgamma 1.2 2025-07-02 [1] RSPM (R 4.6.0)
#> IRanges 2.46.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> irlba 2.3.7 2026-01-30 [1] RSPM (R 4.6.0)
#> iterators 1.0.14 2022-02-05 [1] RSPM (R 4.6.0)
#> jquerylib 0.1.4 2021-04-26 [2] RSPM (R 4.6.0)
#> jsonlite 2.0.0 2025-03-27 [2] RSPM (R 4.6.0)
#> knitr 1.51 2025-12-20 [2] RSPM (R 4.6.0)
#> labeling 0.4.3 2023-08-29 [1] RSPM (R 4.6.0)
#> lattice 0.22-9 2026-02-09 [3] CRAN (R 4.6.1)
#> lazyeval 0.2.3 2026-04-04 [1] RSPM (R 4.6.0)
#> lifecycle 1.0.5 2026-01-08 [2] RSPM (R 4.6.0)
#> listenv 1.0.0 2026-06-22 [1] RSPM (R 4.6.0)
#> locfit 1.5-9.12 2025-03-05 [1] RSPM (R 4.6.0)
#> maaslin3 1.4.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> magrittr 2.0.5 2026-04-04 [2] RSPM (R 4.6.0)
#> MASS 7.3-66 2026-07-15 [3] RSPM (R 4.6.0)
#> Matrix 1.7-6 2026-07-25 [3] RSPM (R 4.6.0)
#> MatrixGenerics 1.24.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> matrixStats 1.5.0 2025-01-07 [1] RSPM (R 4.6.0)
#> memoise 2.0.1 2021-11-26 [2] RSPM (R 4.6.0)
#> mgcv 1.9-4 2025-11-07 [3] CRAN (R 4.6.1)
#> mia 1.20.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> microbiome 1.34.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> mixsqp 0.3-54 2023-12-20 [1] RSPM (R 4.6.0)
#> MultiAssayExperiment 1.38.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> multtest 2.68.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> mvtnorm 1.4-2 2026-07-12 [1] RSPM (R 4.6.0)
#> nlme 3.1-170 2026-07-15 [3] RSPM (R 4.6.0)
#> numDeriv 2016.8-1.1 2019-06-06 [1] RSPM (R 4.6.0)
#> optparse 1.8.2 2026-04-17 [1] RSPM (R 4.6.0)
#> otel 0.2.0 2025-08-29 [2] RSPM (R 4.6.0)
#> parallelly 1.48.0 2026-06-29 [1] RSPM (R 4.6.0)
#> permute 0.9-10 2026-02-06 [1] RSPM (R 4.6.0)
#> phyloseq 1.56.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> pillar 1.11.1 2025-09-17 [2] RSPM (R 4.6.0)
#> pkgbuild 1.4.8 2025-05-26 [2] RSPM (R 4.6.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.6.0)
#> pkgdown 2.2.1 2026-07-07 [2] RSPM (R 4.6.0)
#> pkgload 1.5.3 2026-06-15 [2] RSPM (R 4.6.0)
#> plotly 4.12.1 2026-07-22 [1] RSPM (R 4.6.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.6.0)
#> purrr 1.2.2 2026-04-10 [2] RSPM (R 4.6.0)
#> R6 2.6.1 2025-02-15 [2] RSPM (R 4.6.0)
#> ragg 1.5.2 2026-03-23 [2] RSPM (R 4.6.0)
#> rappdirs 0.3.4 2026-01-17 [2] RSPM (R 4.6.0)
#> RColorBrewer 1.1-3 2022-04-03 [1] RSPM (R 4.6.0)
#> Rcpp 1.1.2 2026-07-05 [2] RSPM (R 4.6.0)
#> registry 0.5-1 2019-03-05 [1] RSPM (R 4.6.0)
#> reshape2 1.4.5 2025-11-12 [1] RSPM (R 4.6.0)
#> rlang 1.3.0 2026-07-05 [2] RSPM (R 4.6.0)
#> rmarkdown 2.31 2026-03-26 [2] RSPM (R 4.6.0)
#> rsvd 1.0.5 2021-04-16 [1] RSPM (R 4.6.0)
#> Rtsne 0.17 2023-12-07 [1] RSPM (R 4.6.0)
#> S4Arrays 1.12.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> S4Vectors 0.50.1 2026-05-13 [1] Bioconductor 3.23 (R 4.6.1)
#> S7 0.2.2 2026-04-22 [1] CRAN (R 4.6.1)
#> sass 0.4.10 2025-04-11 [2] RSPM (R 4.6.0)
#> ScaledMatrix 1.20.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> scales 1.4.0 2025-04-24 [1] RSPM (R 4.6.0)
#> scater 1.40.2 2026-07-02 [1] Bioconductor 3.23 (R 4.6.1)
#> scuttle 1.22.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> Seqinfo 1.2.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> seriation 1.5.8 2025-08-20 [1] RSPM (R 4.6.0)
#> sessioninfo 1.2.4 2026-06-04 [2] RSPM (R 4.6.0)
#> SingleCellExperiment 1.34.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> SparseArray 1.12.2 2026-05-01 [1] Bioconductor 3.23 (R 4.6.1)
#> sparseMatrixStats 1.24.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> SQUAREM 2026.1 2026-03-12 [1] RSPM (R 4.6.0)
#> stringi 1.8.7 2025-03-27 [2] RSPM (R 4.6.0)
#> stringr 1.6.0 2025-11-04 [1] RSPM (R 4.6.0)
#> SummarizedExperiment 1.42.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> survival 3.8-9 2026-07-08 [3] RSPM (R 4.6.0)
#> systemfonts 1.3.2 2026-03-05 [2] RSPM (R 4.6.0)
#> textshaping 1.0.5 2026-03-06 [2] RSPM (R 4.6.0)
#> tibble 3.3.1 2026-01-11 [2] RSPM (R 4.6.0)
#> tidyr 1.3.2 2025-12-19 [1] RSPM (R 4.6.0)
#> tidyselect 1.2.1 2024-03-11 [1] RSPM (R 4.6.0)
#> tidytree 0.4.8 2026-07-02 [1] RSPM (R 4.6.0)
#> treeio 1.36.1 2026-04-30 [1] Bioconductor 3.23 (R 4.6.1)
#> TreeSummarizedExperiment 2.20.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> truncnorm 1.0-9 2023-03-20 [1] RSPM (R 4.6.0)
#> TSP 1.2.7 2026-03-23 [1] RSPM (R 4.6.0)
#> UpSetR 1.4.1 2026-05-25 [1] RSPM (R 4.6.0)
#> usethis 3.2.1 2025-09-06 [2] RSPM (R 4.6.0)
#> utf8 1.2.6 2025-06-08 [2] RSPM (R 4.6.0)
#> vctrs 0.7.3 2026-04-11 [2] RSPM (R 4.6.0)
#> vegan 2.7-5 2026-05-25 [1] RSPM (R 4.6.0)
#> vipor 0.4.7 2023-12-18 [1] RSPM (R 4.6.0)
#> viridis 0.6.5 2024-01-29 [1] RSPM (R 4.6.0)
#> viridisLite 0.4.3 2026-02-04 [1] RSPM (R 4.6.0)
#> webshot 0.5.5 2023-06-26 [1] RSPM (R 4.6.0)
#> withr 3.0.3 2026-06-19 [2] RSPM (R 4.6.0)
#> xfun 0.60 2026-07-09 [2] RSPM (R 4.6.0)
#> XVector 0.52.0 2026-04-28 [1] Bioconductor 3.23 (R 4.6.1)
#> yaml 2.3.12 2025-12-10 [2] RSPM (R 4.6.0)
#> yulab.utils 0.2.4 2026-02-02 [1] RSPM (R 4.6.0)
#>
#> [1] /__w/_temp/Library
#> [2] /usr/local/lib/R/site-library
#> [3] /usr/local/lib/R/library
#> * ── Packages attached to the search path.
#>
#> ──────────────────────────────────────────────────────────────────────────────